Chemistry 251 Laboratory -- Spring 2002
Ibuprofen Project Home Page
Go to Lab Syllabus
Updated 4/19/02
Index
Synthesis of Ibuprofen
Faculty Mentor: John Hanson
Reference: Cleij, M.; Archelas, A.; Furstoss, R. J. Org. Chem. 1999, 64, 5029-5035.
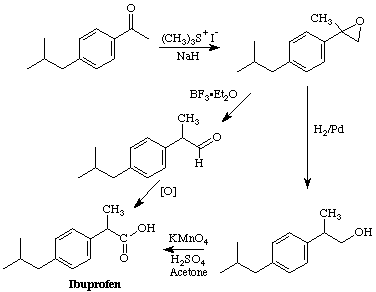
Ibuprofen is the active ingredient in a number of over the counter pain relievers, e.g. Advil, Motrin, and Nuprin. It is one of the top-ten drugs sold worldwide, and, although it has been shown that only the S enantiomer has the desired biological activity, it is currently sold as the racemate. Our synthesis of the racemate, begins with reaction of 4-isobutylacetophenone with the sulfur ylide produced by deprotonation of trimethylsulfonium iodide to yield an epoxide. (You should look this reaction up in an advanced organic text, e.g. the one by Jerry March.) In the JOC article listed above they found that the epoxide could be converted to Ibuprofen by reduction to the alcohol using H2/Pd followed by oxidation of the resulting alcohol to the acid using KMnO4. We found that BF3Et2O catalyzed rearrangement of the epoxide to an aldehyde worked well. Some preliminary attempts at oxidizing this aldehyde to Ibuprofen have been explored.
Students Working on This Project
| Lab Day |
Name |
E-mail |
| Monday |
Gretchen Heinzen |
gheinzen@ups.edu |
| Monday |
Stacy Muffly |
smuffly@ups.edu |
| Tuesday |
Amy Kiesselbach |
akiesselbach@ups.edu |
| Tuesday |
Staci Milnes |
smilnes@ups.edu |
| Wednesday |
Lilian Chan |
lil@dork.com |
| Wednesday |
Aubrey Hendricks |
ahendricks@ups.edu |
| Thursday Aft. |
Paul Tourville |
gotpaul@hotmail.com |
| Thursday Aft. |
Tim Sweeney |
phlewyd@hotmail.com |
| Thursday Eve. |
Danylle Oldis |
doldis@ups.edu |
| Thursday Eve. |
David Anderson |
daanderson@ups.edu |
Table of Reagents and Amounts Available for this Project
The table below lists the chemicals that we will have available for this project. If you need something that is not on this list, consult with the mentor for your project. Also note the "Amount/group" column. This is the total amount of material available for each group to use on the project.
| Reagent |
Source |
Amount/group |
Location |
Comments |
| 4-isobutylacetophenone |
Lancaster
Cat. # 6284 |
5 g |
TA room |
|
| Trimethylsulfonium iodide |
Aldrich
Cat. #T8,048-9 |
4 g |
TA room |
Irritant |
| Sodium Hydride (60% dispersion in mineral oil) |
Aldrich
Cat. #45,291-2 |
5 g |
TA room |
Flammable Solid, Moisture Sensitive |
DMSO (Dimethyl sulfoxide)
Anhydrous |
Aldrich
Cat. #27,685-5 |
100 mL |
TA room |
Wear gloves when handling DMSO. It is readily absorbed through the skin! |
| Boron trifluoride diethyl etherate |
Aldrich
Cat. #21,660-7 |
10 mL |
TA room |
Caution: Toxic, corrosive, moisture sensitive. Dispense under nitrogen in the hood. |
| Palladium powder |
Aldrich
Cat. #32,666-6 |
0.4 g |
TA room |
|
| Sodium chlorite, tech. 80% |
Aldrich
Cat. #24,415-5 |
5 g |
TA room |
|
| Hydrogen peroxide 30% |
Fisher
Cat. #H325-500 |
10 mL |
Cold room (on the left, on the floor) |
Oxidizer. Wear gloves! |
Step 1: Epoxide Formation using Trimethylsulfonium Iodide
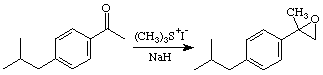
When developing your procedure for Step 1, look at the reports from last year's students. (They are available in a binder in the chemistry library.) Here are some notes on this reaction from Spring 2000:
- The NaH comes as a dispersion in oil. It looks like a grey powder. Be sure not to expose it to air for any significant amount of time. NaH is usually washed with hexane to remove the oil before it is used. Here is a wash procedure that I used when I was doing a reaction that required 2 g of NaH (it was also a 60% dispersion in oil).
- A 100 mL pear shaped flask, equipped with a stir bar was flame dried and cooled under nitrogen. The appropriate amount of NaH was weighed into the flask. The NaH was washed 4 x 10 mL with anhydrous hexane. To do the washing, I added the hexane by syringe and then stirred for a few minutes under Nitrogen. I then turned of the stir bar and let the NaH settle. I then removed most of the hexane with a syringe and repeated the process. After the last wash I removed the remaining hexane by placing on a high vacuum pump, but this might not be necessary.
- Since the NaH is a 60% dispersion in oil, you will have to weigh out 1.67 times the amount you would need if it was pure.
- Is it possible to use NaHMDS instead of NaH? This worked OK, but still some starting material. Let's stick with the NaH.
- Sarah and Jayme got BEAUTIFUL results using 1.5 eq. of NaH, 1.2 eq. of the trimethylsulfonium iodide. They washed the NaH once with hexane, then did the reaction normally. After workup the NMR looked perfect -- no starting material present. No need to do any purification! Click here to see the NMR.
- 4/4/02 -- Amy and Staci had great results, so here is their procedure:
- Preparation of 4'-isobutyl-a-methylstyrene.
A 100-mL pear shaped flask was flame dried and cooled under nitrogen. A solution of 1.03 g of 60% NaH (0.0429 mol) was added to the flask and washed twice with 10 mL of hexane to remove the oil from it. The excess was removed with a syringe. About 15 mL of DMSO was added to the flask and another 15 mL of DMSO was mixed with 3.52 g of (CH3)3S+I- (0.0172 mol). This was also added to the flask and the solution was stirred for roughly 20 minutes at room temperature. Then 2.61 mL of 4-isobutylacetophenone (0.0142 mol) was added dropwise very slowly (over a period of about 10 minutes). The mixture was stirred for 44 hours. It was then worked up by adding 50 mL of water and extraction by diethyl ether, twice with 25-mL portions. The combined ether extracts was back washed with 10 mL of water and again with 10 mL of saturated NaCl. It was then dried with Na2SO4 and put on the rotary evaporator with subsequent analysis by H-NMR and IR. The yield was 90.7% (2.45g, 0.0128 mol). The H-NMR showed peaks at: 0.9 (d, 6H, J=6.59), 1.7 (s, 3H), 1.9 (m, 1H), 2.5 (d, 2H, J=7.14), 2.8 (d, 1H, J=5.49), 3.0 (d, 1H, J=5.22), 7.1 (d, 2H, J=8.24), & 7.3 (d, 2H, J=8.24). The IR shows peaks at: 3030 (sp2 C-H�s), 2960, 2916, 2863 (sp3 C-H�s) and 1511 cm-1 (C=C).
- 4/8/02 -- Danylle and David tried this reaction without washing the NaH with hexanes. It would certainly be much more convenient if we could omit the hexane washes. Unfortunately, the crude product, after aqueous workup, shows very little product. There is mostly starting material with some other unidentified impurities.
Step 2: BF3 Catalyzed Rearrangement
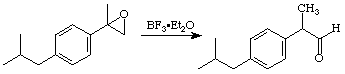
Reference: Garin, D.L.; Gamber, M.; Rowe, B.J. "Epoxidation of Alpha-Methylstyrene and Its Lewis Acid Rearrangement to 2-Phenylpropanal" J. Chem. Educ. 1996, 73, 555.
Notes and Suggestions
- 4/4/02 -- Amy and Staci had great results with this rearrangement. I will try to get a procedure and NMR from them to put up on the web.
- 4/5/02 -- NMR of the aldehyde after the aqueous workup. (Courtesy of Amy and Staci.)
- 4/7/02 -- Here is the procedure from Amy and Staci
- Preparation of 2-(4-isobutylphenyl)-propionaldehyde.
In a 50-mL round bottom flask, 0.51g (0.0036 mol) of 4-isobutyl-a-methylstyrene oxide
was dissolved in 10 mL of dichloromethane. To
this solution, 0.23 mL (0.0018 mol) of boron trifluoride etherate was
added along with a stir bar and the mixture was stirred for 50 minutes.
Another 10 mL of CH2Cl2 was added and the solution was
washed twice with 10-mL portions of saturated NaHCO3 (aq)
and once with 10 mL of saturated NaCl (aq). After drying
the mixture with Na2SO4, it was put in the rotorary evaporator followed by analysis by H-NMR and IR. The yield was 67% (0.33 g, 0.0017 mol). NMR
Step 3: Oxidation of the Aldehyde to the Acid
Notes and Suggestions
- 4/4/02 -- We need to get some procedures for this. I know some of you found some references when you did your initial Beilstein searches. Please bring them by so we can look at them!
- 4/7/02 -- Amy Kiesselbach and I did a Beilstein Commander search and came up with an possible procedure for the oxidation of the aldehyde to the acid. This procedure is for oxidation of hydratropaldehyde to hydratropic acid. These molecules are similar to the ones we are working with, but lack the isobutyl group on the aromatic ring. You will need to scale down this procedure considerably.
- Reference: Ebbers, E.J.; Ariaans, G.J.A.; Bruggink, A.; Zwanenburg, B. "Controlled racemization and asymmetric transformation of a-substituted carboxylic acids in the melt" Tetrahedron: Asymmetry 1999, 10, 3701-3718.
- Hydratropic acid. A solution of NaClO2 (179.4 g, 1.6 mol) in water (750 mL) was added dropwise over a period of 3 h to a stirred mixture of hydratropaldehyde 18 (150.5 g, 1.1 mol) in acetonitrile (750 ml) and NaH2PO4xH2O (35.9 g, 0.26 mol) in water (300 ml) and 35 % hydrogen peroxide (112 ml, 1.3 mol) while keeping the temperature at 0-10 degrees C. Oxygen evolved from the solution until the end of the reaction (3 h). To the stirred solution was added Na2SO3 (20 g) to destroy unreacted HOCl and H2O2, followed by the addition of NaHCO3 until pH 9-10. The resulting mixture was extracted with dichloromethane (three times), acidigied with concentrated hydrochloride until pH-1-2 and extracted with dichloromethane (four times). the combined organic layers from the acidic extraction were dried over MgSO4 and concentrated in vacuo to give pure hydratropic acid as an oil (154.3 g, 91%).
- 4/7/02 -- Amy and I also found another promising reference that did the same oxidation in 98 percent yield. They also used NaClO2 in a mixture of dichloromethane/acetic acid. But we will need to order the reference. We'll also need help from someone who can read French!
- Reference: Bayle, J.P.; Perez, F.; Courtieu, J. Bull. Soc. Chim. Fr. 1990, 4, 565-567.
- 4/15/02 -- Amy, Staci, Danylle, and David have tried this procedure. Amy's preliminary results looked like a mixture of compounds, but it may contain some of the desired product.
- 4/15/02 -- Gretchen and Stacy provided me with another procedure, again using sodium chlorite, that we might want to try.
- Reference: Babu, B.R.; Balasubramaniam, K.K. "Simple and Facile Oxidation of Aldehydes to Carboxylic Acids" Org. Prep. Proced. Intl. 1994, 26, 123-125.
- General Procedure for Oxidation of Aldehydes. - A solution (0.10-0.14 mole) of sodium chlorite in 100 mL of water was added dropwise in one hr to a stirred mixture of aldehyde (0.10 mol) (99% purity) in 50-100 mL of acetonitrile. Temperature was maintained at 10 degrees or below for 2 hrs and the reaction mixture was then stirred at room temperature for another 3 hrs. then 10% sodium hydroxide solution (100 mL) was added and the resulting reaction mixture was extracted with ether (3 x 50 mL). The separated aqueous solution was cooled in an ice-bath and acidified (pH 2-3) with cold dilute hydrochloric acid. the separated solid was collected, dried and recrystallized from a suitable solvent to give the pure carboxylic acid (TLC and GC).
- Other references for oxidation of aldehydes using sodium chlorite are listed in the Fluka catalog: "Reagent in combination with sulfamic acid, for a convenient oxidation of aldehydes to carboxylic acids" Lindgeren, B.O.; Nilsson, T. Acta Chem. Scand. 1973, 27, 888. Colombo, L., et. al., J. Chem. Soc. Perkin I 1980, 136.
- 4/16/02 -- I did some looking in Fieser and Fieser and in the Encyclopedia of Organic Reagents and found some more references. I learned that the rest of the 80% sodium chlorite is NaCl. I also found a good general reference to the use of sodium chlorite for the oxidation of aldehydes: Reference: Dalcanale, E.; Montanari, F. "Selective Oxidation of Aldehydes to Carboxylic Acids with Sodium Chlorite-Hydrogen Peroxide" J. Org. Chem. 1986, 51, 567-569. Their procedure is similar to that described in the Ebbers et. al reference above. But I recommend looking at this reference for more information.
- 4/18/02 -- Danylle and David's product looked great by NMR, but they said the yield was only about 3.5%. Here is their procedure:
- In a 25 mL RB flask, aldehyde (0.25 g, 1.4 mmol), acetonitrile (0.5 mL, 9.19
mmol), NaH2PO4 (0.5 g, 0.3 mmol) and 30% hydrogen peroxide (0.144 mL, 1.65
mmol) were stirred at 0-10 degrees C. NaClO2 (0.18 g, 2.03 mmols) dissolved in 1 mL
water was added dropwise over 1 hour. During the hour, oxygen gas evolved off
of solution which was stirred for 20 minutes after the 1 hr it took to add
NaCl02 (after this, no more oxygen bubbles formed). Na2SO3 (0.03 g) was aded
and NaHCO3 was added until pH=9-10. Wash 2x with dichloromethane and add HCl
to combined washes until pH=1-2. Extract with 4 washes of dichloromethane.
Dry with NaSO4, filter, roto-vap.
H-NMR: DAA050.
- 4/18/02 -- Amy got a copy of the Bull. Soc. Chim. Fr. article ("Oxydation des aldehydes par le chlorite de sodium") referred to above. We will try to get relavent parts translated.
- 4/19/02 -- Danylle and David tried the oxidation reaction using essentially the same procedure as last time. They started with 250 mg of starting material and ended up with about 10 mg of material whose NMR was pretty ugly. They also looked at their initial dichloromethane washes (before acidification) and they recovered 40 mg of material that also didn't look very good by NMR. I'm not sure why this reaction is not working better. Maybe the French method will work better?
Alternative Step 2: H2/Pd Opening of Epoxide
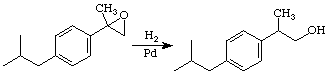
Reference: Cleij, M.; Archelas, A.; Furstoss, R. J. Org. Chem. 1999, 64, 5029-5035.
Notes and Suggestions
- 4/5/02 -- Paul and Tim are trying this procedure. They substituted DMF for N-methylformamide, and didn't bother to do it at 0 degrees.